42 research outputs found
Rotavirus Rearranged Genomic RNA Segments Are Preferentially Packaged into Viruses Despite Not Conferring Selective Growth Advantage to Viruses
 Get PDF
Get PDFThe rotavirus (RV) genome consists of 11 double-stranded RNA segments. Sometimes, partial sequence duplication of an RNA segment leads to a rearranged RNA segment. To specify the impact of rearrangement, the replication efficiencies of human RV with rearranged segments 7, 11 or both were compared to these of the homologous human wild-type RV (wt-RV) and of the bovine wt-RV strain RF. As judged by viral growth curves, rotaviruses with a rearranged genome (r-RV) had no selective growth advantage over the homologous wt-RV. In contrast, r-RV were selected over wt-RV during competitive experiments (i.e mixed infections between r-RV and wt-RV followed by serial passages in cell culture). Moreover, when competitive experiments were performed between a human r-RV and the bovine wt-RV strain RF, which had a clear growth advantage, rearranged segments 7, 11 or both always segregated in viral progenies even when performing mixed infections at an MOI ratio of 1 r-RV to 100 wt-RV. Lastly, bovine reassortant viruses that had inherited a rearranged segment 7 from human r-RV were generated. Although substitution of wt by rearranged segment 7 did not result in any growth advantage, the rearranged segment was selected in the viral progenies resulting from mixed infections by bovine reassortant r-RV and wt-RV, even for an MOI ratio of 1 r-RV to 107 wt-RV. Lack of selective growth advantage of r-RV over wt-RV in cell culture suggests a mechanism of preferential packaging of the rearranged segments over their standard counterparts in the viral progeny
Réarrangements génomiques et génétique inverse des rotavirus
 No full text
No full textLes rotavirus (RV) sont les principaux agents des gastro-entérites aigües du nourrisson. Leur génome est composé de 11 segments d ARN bicaténaire. Le réarrangement génomique est l un des mécanismes de la variabilité génétique des RV. Cet événement rare consiste en une duplication partielle de la séquence d un segment suite à une recombinaison intra-moléculaire. Les conséquences des réarrangements génomiques sur la capacité réplicative de RV à génome réarrangé n ont été que peu étudiées. Nous avons montré que les segments réarrangés 7 et/ou 11 ne confèrent pas d avantage réplicatif aux RV humains par rapport à leur homologue sauvage. Paradoxalement, les segments réarrangés sont ségrégés préférentiellement dans la descendance virale lors d expériences de compétition entre un RV à génome réarrangé et un RV sauvage. L ensemble de ces résultats suggère que les segments réarrangés 7 et/ou 11 des RV humains sont encapsidés préférentiellement à leur homologue sauvage lors de la réplication virale. Nous avons ensuite développé un système de génétique inverse pour les RV basé sur l utilisation de l encapsidation préférentielle des segments réarrangés. Ce système qui utilise comme exogène un segment réarrangé et comme virus auxiliaire un RV bovin a permis l obtention des RV recombinants ayant intégré un segment 7 réarrangé modifié in vitro, notamment un RV recombinant qui exprime une protéine NSP3 modifiée d un poids moléculaire correspondant au double de celui de la protéine sauvage. Enfin, des expériences complémentaires ont été initiées afin de généraliser le système à d autres segments réarrangés ou à d autres virus auxiliairesPARIS-BIUSJ-Physique recherche (751052113) / SudocSudocFranceF
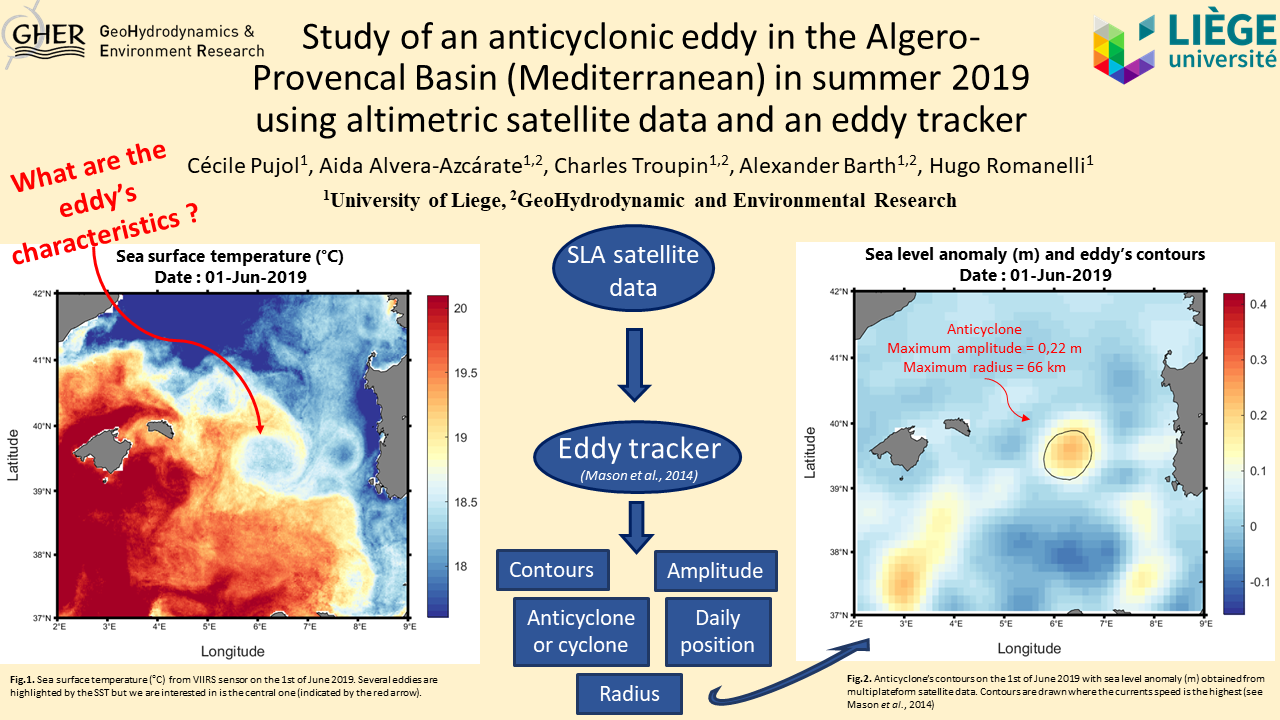
Etudy d'un tourbillon anticyclonique dans le bassin Algéro-Provençal (Mer Méditerranée) observé lors de l'été 2019 en utilisant des données altimétriques et un eddy tracker
 Full text link
Full text linkIn April 2019, a large anticyclonic Eddy has formed in Western Mediterranean Sea between Sardinia and Balearic Islands. This anticyclone was observable with Sentinel-3 SST satellite data for 7 months and its diameter was estimated to 150 km. Although mesoscale anticyclones are quite common in this part of the Mediterranean Sea, such large and long-live eddies remain exceptional and repercussions for ocean-atmospheric exchanges and for biodiversity might be consequent. However, due to the increase of temperatures during summer, the satellite SST track of the eddy has been lost during a few weeks in August and September. Indeed, the SST signature of the eddy was not distinguishable from surrounding waters anymore. In order to track the eddy during its entire life and have a better understanding of its characteristics, sea level anomaly derived from altimetric data will be analysed in this study with the Py Eddy Tracker toolbox to investigate the variation of its position, its altimetry and its size. The distribution of other remarkable eddies in this zone and period will also be considered. Moreover, a high-resolution SST field will be reconstructed with DINEOF method so the comparison between eddy’s SST and altimetric characteristics will be assured
Dual Combined Real-Time Reverse Transcription Polymerase Chain Reaction Assay for the Diagnosis of Lyssavirus Infection
 No full text
No full textInternational audienceThe definitive diagnosis of lyssavirus infection (including rabies) in animals and humans is based on laboratory confirmation. The reference techniques for post-mortem rabies diagnosis are still based on direct immunofluorescence and virus isolation, but molecular techniques, such as polymerase chain reaction (PCR) based methods, are increasingly being used and now constitute the principal tools for diagnosing rabies in humans and for epidemiological analyses. However, it remains a key challenge to obtain relevant specificity and sensitivity with these techniques while ensuring that the genetic diversity of lyssaviruses does not compromise detection. We developed a dual combined real-time reverse transcription polymerase chain reaction (combo RT-qPCR) method for pan-lyssavirus detection. This method is based on two complementary technologies: a probe-based (TaqMan) RT-qPCR for detecting the RABV species (pan-RABV RT-qPCR) and a second reaction using an intercalating dye (SYBR Green) to detect other lyssavirus species (pan-lyssa RT-qPCR). The performance parameters of this combined assay were evaluated with a large panel of primary animal samples covering almost all the genetic variability encountered at the viral species level, and they extended to almost all lyssavirus species characterized to date. This method was also evaluated for the diagnosis of human rabies on 211 biological samples (positive n = 76 and negative n = 135) including saliva, skin and brain biopsies. It detected all 41 human cases of rabies tested and confirmed the sensitivity and the interest of skin biopsy (91.5%) and saliva (54%) samples for intra-vitam diagnosis of human rabies. Finally, this method was successfully implemented in two rabies reference laboratories in enzootic countries (Cambodia and Morocco). This combined RT-qPCR method constitutes a relevant, useful, validated tool for the diagnosis of rabies in both humans and animals, and represents a promising tool for lyssavirus surveillance
Customized online and onsite training for rabies-control officers.
 No full text
No full textInternational audienceIt is difficult to deliver adequate training for people working in rabies control in low and middle-income countries. Popular e-learning systems for low-income settings are not well suited to developing and testing practical skills, including laboratory methods.We customized training in rabies control methods for African professionals and students from different disciplines. Trainees participated in preparatory online sessions, evaluations and exercises before and after a 12-day workshop. Trainees and mentors continued to interact through an online forum up to one year after the workshop.In Africa, 15,000 deaths from rabies occur each year due to a lack of awareness, inaccessibility of post-exposure prophylaxis, inadequate or absent canine rabies-control programmes and lack of governmental financial support.Thirty two trainees - working in health departments, hospitals, veterinary stations and research institutes - were selected to participate; 28 completed the course and passed the final evaluation. Pilot rabies investigation programmes were developed, and two manuscripts submitted for publication. An online forum facilitated further progress for a year after the workshop.A combination of customized online and onsite training is suitable for teaching disease-control personnel in low-income countries. Participation in this course enabled trainees to advocate for the development of national disease-control strategies. Mentoring is needed to develop a strong network of experts in similar settings
Using phylogeographic approaches to analyse the dispersal history, velocity and direction of viral lineages — Application to rabies virus spread in Iran
 No full text
No full textinfo:eu-repo/semantics/publishe
Comparison of intra- and inter-host genetic diversity in rabies virus during experimental cross-species transmission
 No full text
No full textInternational audienceThe development of high-throughput genome sequencing enables accurate measurements of levels of sub-consensus intra-host virus genetic diversity and analysis of the role played by natural selection during cross-species transmission. We analysed the natural and experimental evolution of rabies virus (RABV), an important example of a virus that is able to make multiple host jumps. In particular, we (i) analyzed RABV evolution during experimental host switching with the goal of identifying possible genetic markers of host adaptation, (ii) compared the mutational changes observed during passage with those observed in natura, and (iii) determined whether the colonization of new hosts or tissues requires adaptive evolution in the virus. To address these aims, animal infection models (dog and fox) and primary cell culture models (embryo brain cells of dog and fox) were developed and viral variation was studied in detail through deep genome sequencing. Our analysis revealed a strong unidirectional host evolutionary effect, as dog-adapted rabies virus was able to replicate in fox and fox cells relatively easily, while dogs or neuronal dog cells were not easily susceptible to fox adapted-RABV. This suggests that dog RABV may be able to adapt to some hosts more easily than other host variants, or that when RABV switched from dogs to red foxes it lost its ability to adapt easily to other species. Although no difference in patterns of mutation variation between different host organs was observed, mutations were common following both in vitro and in vivo passage. However, only a small number of these mutations also appeared in natura, suggesting that adaptation during successful cross-species virus transmission is a complex, multifactorial evolutionary process
Large-Scale Phylogenomic Analysis Reveals the Complex Evolutionary History of Rabies Virus in Multiple Carnivore Hosts.
 Get PDF
Get PDFInternational audienceThe natural evolution of rabies virus (RABV) provides a potent example of multiple host shifts and an important opportunity to determine the mechanisms that underpin viral emergence. Using 321 genome sequences spanning an unprecedented diversity of RABV, we compared evolutionary rates and selection pressures in viruses sampled from multiple primary host shifts that occurred on various continents. Two major phylogenetic groups, bat-related RABV and dog-related RABV, experiencing markedly different evolutionary dynamics were identified. While no correlation between time and genetic divergence was found in bat-related RABV, the evolution of dog-related RABV followed a generally clock-like structure, although with a relatively low evolutionary rate. Subsequent molecular clock dating indicated that dog-related RABV likely underwent a rapid global spread following the intensification of intercontinental trade starting in the 15th century. Strikingly, although dog RABV has jumped to various wildlife species from the order Carnivora, we found no clear evidence that these host-jumping events involved adaptive evolution, with RABV instead characterized by strong purifying selection, suggesting that ecological processes also play an important role in shaping patterns of emergence. However, specific amino acid changes were associated with the parallel emergence of RABV in ferret-badgers in Asia, and some host shifts were associated with increases in evolutionary rate, particularly in the ferret-badger and mongoose, implying that changes in host species can have important impacts on evolutionary dynamics
Geographic Disparities in Domestic Pig Population Exposure to Ebola Viruses, Guinea, 2017–2019
 No full text
No full textInternational audienceAlthough pigs are naturally susceptible to Reston virus and experimentally to Ebola virus (EBOV), their role in Orthoebolavirus ecology remains unknown. We tested 888 serum samples collected from pigs in Guinea during 2017–2019 (between the 2013–16 epidemic and its resurgence in 2021) by indirect ELISA against the EBOV nucleoprotein. We identified 2 hotspots of possible pig exposure by IgG titer levels: the northern coast had 48.7% of positive serum samples (37/76), and Forest Guinea, bordering Sierra Leone and Liberia, where the virus emerged and reemerged, had 50% of positive serum samples (98/196). The multitarget Luminex approach confirms ELISA results against Ebola nucleoprotein and highlights cross-reactivities to glycoprotein of EBOV, Reston virus, and Bundibugyo virus. Those results are consistent with previous observations of the circulation of Orthoebolavirus species in pig farming regions in Sierra Leone and Ghana, suggesting potential risk for Ebola virus disease in humans, especially in Forest Guinea

